Синдром Пфайффер: причини, симптоми, лікування
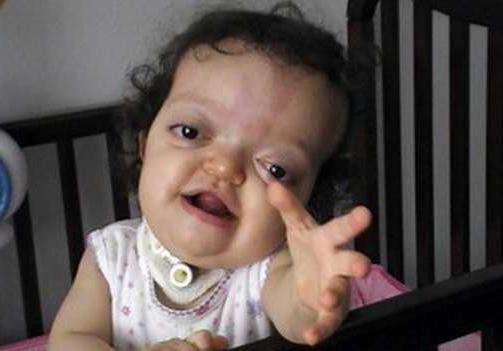
Синдром Пфайффер - надзвичайно рідкіснегенетичне захворювання, яке зустрічається в середньому у одного з 100 тис. новонароджених. Однаково часто вражає хлопчиків і дівчаток. Основний симптом хвороби - раннє зрощення кісток черепа в процесі ембріогенезу, через що мозок не може в подальшому нормально розвиватися.
Загальні відомості
Захворювання було відкрито відомим німецькимгенетиком Рудольфом Пфайффер. У 1964 році він описав симптоми раніше невідомої хвороби, основними ознаками якої були аномалії в структурі черепа і зрощення пальців на ступнях або кистях рук (синдактилія). Синдром спостерігався у восьми пацієнтів з однієї сім'ї, що говорило про генетичну природу хвороби. Подальші дослідження показали, що синдром Пфайффер передається по аутосомно-домінантним типом.
Ультразвукове дослідження дозволяє виявити це захворювання у плода вже в другому триместрі вагітності. На УЗД видно аномалії розвитку черепа, внутрішніх органів і кінцівок.
симптоми
У 1993 році американський генетик Майкл Коензапропонував класифікувати синдром Пфайффер на три типи залежно від тяжкості симптомів. Зараз його класифікація є загальноприйнятою і широко використовується в медичній літературі.
Симптоми синдрому Пфайффер 1 типу включаютькраніосиностозу - стан, при якому один або декілька черепних швів передчасно зливаються, утворюючи цілісну кістка. Як результат відзначаються зміни в формі черепа і аномальні риси обличчя - гипертелоризм, низько посаджені вуха. Може мати місце погіршення зору, підвищений внутрішньочерепний тиск, розщеплення неба. У 50% пацієнтів відзначається зниження слуху. Більшість людей з даним типом хвороби мають нормальним рівнем інтелекту і не мають неврологічних відхилень.
У дітей з типом 1 синдрому Пфайффер зовнішніознаки, такі як широкі короткі пальці на руках і ногах, спостерігаються майже завжди. Синдактилія - можливий, але не обов'язковий симптом для цього типу патології.

Даний тип синдрому успадковується аутосомно-домінантно.
Нижче представлено фото синдрому Пфайффер. Тривалість життя в Росії для людей, які страждають на це захворювання, перевищує 60 років.

Для захворювання 2-го типу характерний череп вформі "трилисника", злиття хребців, синдактилія та зміщення очних яблук вперед (проптоз). З перших днів життю проявляються серйозні неврологічні порушення.
3-й тип захворювання характеризується ще більшраннім Краніосиностоз. Череп аномально витягнутий і не утворює "трилисник". Мають місце аномалії зубів, гипертелоризм, розумова відсталість, пороки розвитку внутрішніх органів.
Тривалість життя при 2-м і 3-м типізахворювання становить від декількох днів до декількох місяців - пороки внутрішніх органів і неврологічні порушення не сумісні з життям. Мутації, що відповідають за ці два типи синдрому Пфайффер, виникають спорадично і генетично не успадковуються.
причини
Синдром Пфайффер пов'язаний з мутаціями рецептора 1фактора росту фібробластів (FGFR1) на хромосомі 8 або з рецептором фактора росту фібробластів 2 (FGFR2) на хромосомі 10. Гени, які зачіпають мутації, відповідають за нормальний ріст фібробластів, що грає ключову роль в розвитку кісток організму.

лікування
Сучасна медицина поки не дозволяє усунутимутації в людському геномі, навіть якщо відомо, в яких саме генах відбулися перебудови. Тому для синдрому Пфайффер, як для більшості інших генетичних захворювань, лікування є симптоматичним. Для пацієнтів з 2-м і 3-м типом захворювання прогноз несприятливий: численні аномалії розвитку не можуть бути скориговані ні медикаментозно, ні хірургічно.
Синдром Пфайффер 1 типу сумісний з життям. Основний симптом захворювання - передчасне зрощення кісток черепа - може бути скоректований хірургічно. Операцію рекомендується проводити дітям до досягнення тримісячного віку.
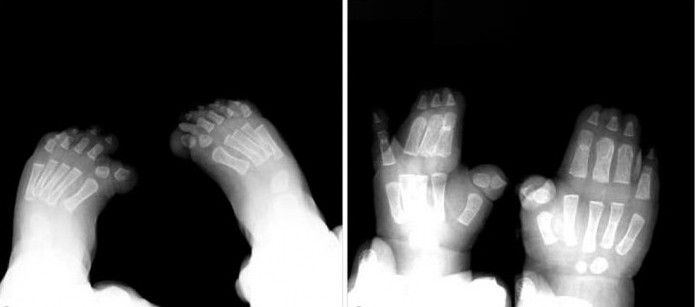
Крім того, хірургічне втручання є ефективним в разі простою синдактилії. Лікування найкраще починати у віці 18-24 місяців, в період активного росту пальців на руках і ногах.